Difference: JaysJournal (68 vs. 69)
Revision 692010-08-11 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 88 to 88 | ||||||||
| ||||||||
| Added: | ||||||||
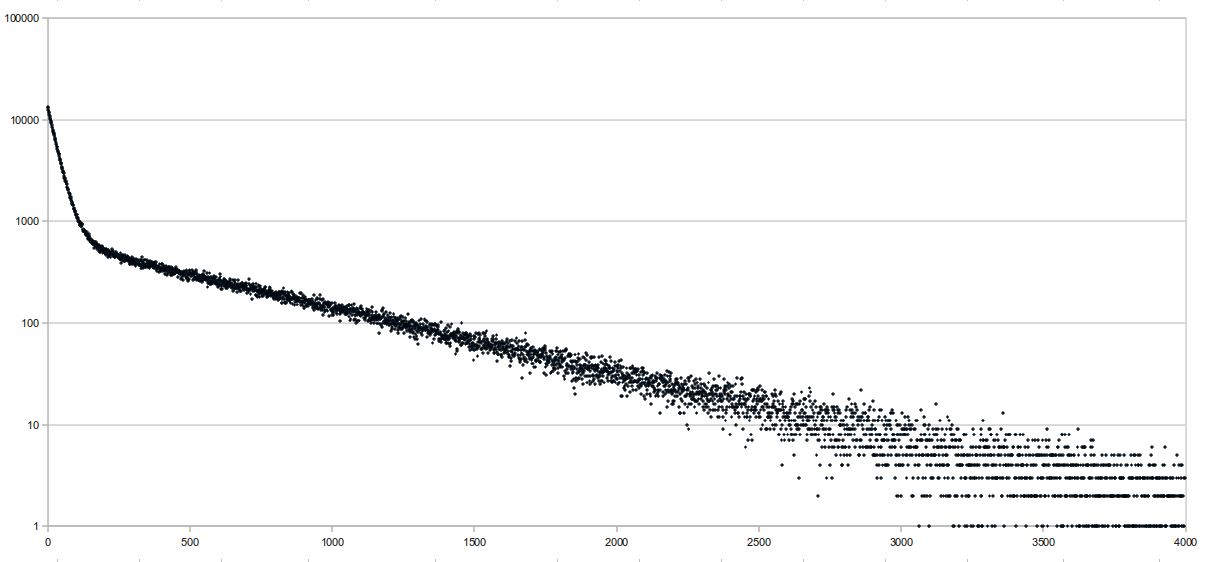
| > > | 08/11/10Implemented a save/load feature for the FM index. I'm also thinking of implementing a "partial" load feature, where only the BWT string is loaded, and the other data structures are still built. The reason for this is that the BWT string is the one that takes the most memory (and maybe time, too) to build and should be constant for all indexes, while the other structures will differ depending on sampling rates and memory restrictions. So, the BWT string can be passed around between machines (with different memory restrictions) easily, while the other data structures can't. I also did a few preliminary benchmarks, and the times were not great on theLocate function. I think this might be because we don't implement Locate the "proper" way, which guarantees a successful locate within sampling rate number of queries. Following Chris' suggestion, I tried graphing the number of backtracks it takes before a successful location is found on a random readset, and here are the results:
The sequence length was 65,751,813 bases long and consisted of the E. coli genome (4.6 Mbp) repeated multiple times. Reads were randomly generated 10-base sequences, and only the first 10 matches were "located". This was run at a sampling rate of 64 bases, and the average distance to locate was 341.68, which isn't very good. The x axis of the graph represents the distance it takes to locate, and the y axis is the number of aligments with that distance (it's logged). There were a total
 To do:
To do:
| |||||||
| ||||||||
| Added: | ||||||||
| > > |
| |||||||
View topic | History: r73 < r72 < r71 < r70 | More topic actions...
Ideas, requests, problems regarding TWiki? Send feedback