May 2010 archive June 2010 archive
July 2010 archive
June 2010 archive
July 2010 archive
07/30/10
Started to implement the new StaticDNASequence. The build process is going to get a little ugly, but the ranks should still be relatively clean (which is what matters). To make it easier on myself, I'm just going to do one pass through the whole sequence first to find all the N's and build the new data structure, then build the rest of the rank structure (rank blocks + count blocks). This is kind of wasteful because I can actually build the $ structure while building the rank structure (for just one pass), but I think it might be harder, given my current code. So, the build process will be a bit slower (which I think is okay...).
Finally, I did some benchmarks on the
count function. Test parameters are:
- read length = 35
- reference = e coli genome NC_010473, minus two characters (there was one Y and one R...) (length = 4,686,136 bases)
- number of reads = 10,000,000, generated randomly
- this randomly generated set was copied 10 times, for a total of 100,000,000 calls to
count.
- random seed = 1234
- times are reported in milliseconds wall time.
- Build version is 64-bit, Release mode (
-O3)
- Memory usage is just the memory used by the rank structure
| Sampling rate (bases) |
Memory usage (bytes) |
Type |
SSE 4.2? |
Time (ms) |
| 64 |
1,784,768 |
2-bit |
No |
27,212 |
| 256 |
1,339,168 |
2-bit |
No |
46,663 |
| 64 |
2,973,048 |
3-bit |
No |
31,454 |
| 256 |
2,081,848 |
3-bit |
No |
52,876 |
| 2048 |
1,821,912 |
3-bit |
No |
156,832 |
| 64 |
1,784,768 |
2-bit |
Yes |
19,924 |
| 256 |
1,339,168 |
2-bit |
Yes |
33,002 |
| 64 |
2,973,048 |
3-bit |
Yes |
24,371 |
| 256 |
2,081,848 |
3-bit |
Yes |
38,524 |
| 2048 |
1,821,912 |
3-bit |
Yes |
90,328 |
For a (very) rough comparison,
saligner, using readaligner's FM index, takes 64.461 seconds to do an exact mapping for 2.7 million reads (each 35 bases in length). This means it would take roughly 2,387 seconds to align 100,000,000 reads. However, this is not a very good comparison because that one's doing a
locate function which is more complex, has to read from a file and write to an output file, and runs at 32 bits. Also, the rank structure might use a different sampling rate, although I believe it uses a sampling rate of 256, which is why I included that test in. So, this means we have quite a bit of room to work with!
Another note is that for the 3-bit structure, it requires a sampling rate of 2048 to be roughly comparable in memory usage. However, at this rate, it's much slower than the 2-bit structure (roughly 5-6 times slower).
To do:
- Finish the $ data structure implementation in the StaticDNASequence
08/04/10
Spent all of yesterday and most of today finishing up the $ data structure for the StaticDNASequence class. I've finished testing it thoroughly (I hope). I've also integrated it with the FM Index and tested the count method to verify the output. Basically, I used the
std::string::find method to look through a reference sequence (I used
NC_010473, the E. coli genome) to "manually" count the occurrences of some read. Then, I used the actual
FMIndex::Count method to count it and verified that they were the same. I tested a bunch of random sequences, and every one of them came out correct. So, I'm pretty sure that my rank structure and count methods are correct.
Also did a few benchmarks using the new 2-bit "enhanced" version and repeated some benchmarks from last time:
Test parameters:
- read length = 35
- reference = e coli genome NC_010473, minus two characters (there was one Y and one R...) (length = 4,686,136 bases)
- number of reads = 100,000, generated randomly
- this randomly generated set was copied 1000 times, for a total of 100,000,000 calls to
count.
- random seed = 1234
- times are reported in milliseconds wall time.
- Build version is 64-bit, Release mode (
-O3)
- Memory usage is just the memory used by the rank structure
- There was one $ in the sequence, no N's
| Sampling rate (bases) |
Memory usage (bytes) |
Type |
SSE 4.2? |
Time (ms) |
Ratio to 2-bit |
| 64 |
1,759,632 |
2-bit |
No |
27.071 |
1 |
| 256 |
1,320,312 |
2-bit |
No |
46,726 |
1 |
| 2048 |
1,192,176 |
2-bit |
No |
137,583 |
1 |
| 64 |
1,761,936 |
2-bit "special" |
No |
27,778 |
0.97 |
| 256 |
1,322,640 |
2-bit "special" |
No |
47,574 |
0.98 |
| 2048 |
1,194,728 |
2-bit "special" |
No |
138,833 |
0.99 |
| 64 |
2,931,176 |
3-bit |
No |
31,513 |
0.86 |
| 256 |
2,052,536 |
3-bit |
No |
53,110 |
0.88 |
| 2048 |
1,796,264 |
3-bit |
No |
156,759 |
0.88 |
| 64 |
1,759,632 |
2-bit |
Yes |
19,747 |
1 |
| 256 |
1,320,312 |
2-bit |
Yes |
32,898 |
1 |
| 2048 |
1,192,176 |
2-bit |
Yes |
74,230 |
1 |
| 64 |
1,761,936 |
2-bit "special" |
Yes |
20,471 |
0.96 |
| 256 |
1,322,640 |
2-bit "special" |
Yes |
33,809 |
0.97 |
| 2048 |
1,194,728 |
2-bit "special" |
Yes |
76,269 |
0.99 |
| 64 |
2,931,176 |
3-bit |
Yes |
24,855 |
0.79 |
| 256 |
2,052,536 |
3-bit |
Yes |
37,847 |
0.87 |
| 2048 |
1,796,264 |
3-bit |
Yes |
89,793 |
0.84 |
To do:
- Start doing a
Locate method.
- Maybe do some code cleanup
08/06/10
Yesterday and today, I did a bunch of cleanup on both the rank structure and FM index classes. I pulled some methods out of
StaticDNAStructure::Init so the code can be cleaner. I also rethought the naming scheme of a lot of variables to make the code a little more readable. For the FM index, I also pulled out methods from the
FMIndex::Init method and overloaded the method to take in C++ strings (which are used by
saligner currently). I also added the building of the BWT string directly into the FM index class instead of making it built externally. Currently, only one sequence is supported, but later on, there will be multi-sequence support.
Chris suggeted in an email that we prefetch
ep ranks while doing both counts and locates, which might make it faster. I'll be discussing this with him later.
Finally, I finished the
FMIndex::Locate function today. It works as expected, and I've tested it pretty thoroughly. Locate requires a bunch of LF mappings of each position in the BWT string to travel "backwards" until a position is reached whose position has been indexed. However, I can't actually get the specific character at the given position in the BWT to do the LF mapping. The FM index paper suggests doing an LF on both position
i and
i-1 for each character in the alphabet, since the only one that will differ in LF value is the character I'm currently on. I'm also thinking I could just implement a
select function in my rank structure, which shouldn't be too hard.
To do:
- Benchmarks! I might be able to integrate the index into
saligner without too much trouble, so we can get some pretty accurate comparisons?
- Implement saving/loading of the whole index. Currently, I support saving/loading of the rank structure, and the FM index is just a few more data structures so it shouldn't be too bad.
08/11/10
Implemented a save/load feature for the FM index. I'm also thinking of implementing a "partial" load feature, where only the BWT string is loaded, and the other data structures are still built. The reason for this is that the BWT string is the one that takes the most memory (and maybe time, too) to build and should be constant for all indexes, while the other structures will differ depending on sampling rates and memory restrictions. So, the BWT string can be passed around between machines (with different memory restrictions) easily, while the other data structures can't.
I also did a few preliminary benchmarks, and the times were not great on the
Locate function. I think this might be because we don't implement
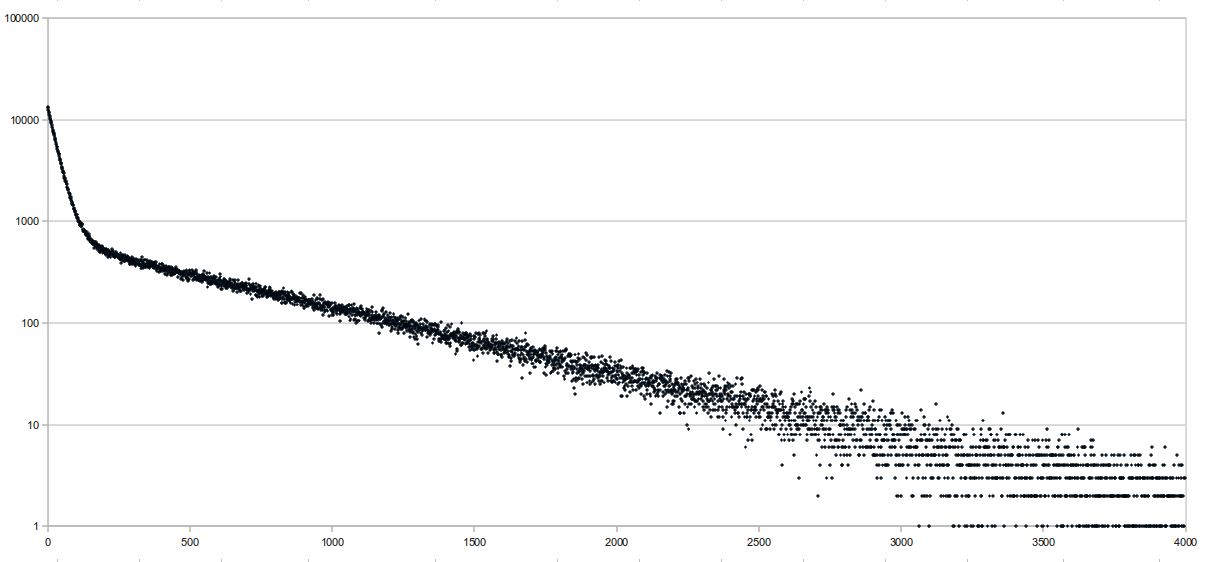
Locate the "proper" way, which guarantees a successful locate within sampling rate number of queries. Following Chris' suggestion, I tried graphing the number of backtracks it takes before a successful location is found on a random readset, and here are the results:
The sequence length was 65,751,813 bases long and consisted of the E. coli genome (4.6 Mbp) repeated multiple times. Reads were randomly generated 10-base sequences, and only the first 10 matches were "located". This was run at a sampling rate of 64 bases, and the average distance to locate was 341.68, which isn't very good. The x axis of the graph represents the distance it takes to locate, and the y axis is the number of aligments with that distance (it's logged). There were a total
- Reference length = 65,751,813 bp (consisting of the E. coli genome repeated multiple times)
- Randomly generated 10-base sequences, only the first 10 alignments were "located"
- Run at a sampling rate of 64 bases for the locate structure
- Average distance = 341.68, which isn't very good
- x-axis is the distance it takes to locate, y-axis is the number of alignments with that distance, semi-logged
- 855,150 locates performed in total
To do:
- Make a "proper" locate structure and compare.
