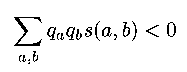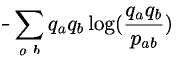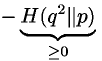- assuming sequence positions are statistically independent (the occurence of a 'letter' in a sequence is independent of the surrounding letters)
- for the ungapped case: (there are no gaps in either sequence)
- Definition: qa and qb denote the probability of generating 'letter' a, b
- We want: (s(a,b) is the score of a,b)
- using the definition of s(a,b) as a log odds score (see lecture 1), we get:
- which can be reduced to
- where H is the relative entropy of distribution q2 with respect to distribution p [Note: relative is a measure of how different two distributions are.]. Relative entropy is always greater or equal to zero (theorem from statistics, see Chapter 11 of 'Biological Sequence Analysis for a proof') and zero only of the two distributions being compared are identical. Hence, our condition is satisfied.
- using these math/statistic theorems, the result is: if we score the way we did, and qa, qb 'behave well', the score of aligning a random subsequence is < 0.
- for the gapped case (more realistic):
- analogous analysis is not possible.
- But since that is extremely relevant in practice, people tweak the matrices until 'they make sense'.
[Note: this assumption is quite unrealistic - nevertheless what follows of it seems to have practical relevance]