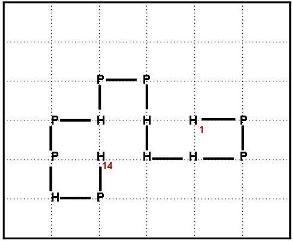
(a) Consider the following structure of the hydrophobic-polar sequence HPPHHHPPHPPHPH in the 2D HP model:
(b) Show all the configurations that could be reached by 1 local search move (end, corner or crankshaft move) from the structure shown in part (a). Calculate the free energy for each structure (configuration) you've found under the HP model. Also, report the acceptance probability for each structure you've found under Metropolis criterion for a given temperature parameter T. [10 marks]
(c) Find an alternate structure for the sequence from part (a) with lower free energy (Hint: you can reach a structure with energy <= -6 through a combination of 2 local search moves) [7 marks]
(d) Briefly discuss in which ways the 2D HP model can be extended in order to obtain a more realistic model. [5 marks]
(a) Briefly explain the role of the hydrophobic force in protein folding. [5 marks]
(b) Briefly explain Levinthal's paradox. [5 marks]
(c) List three fundamental approaches to tertiary protein structure prediction and briefly explain under which circumstances each of them is appropriate. [10 marks]
(a) Given the following RNA secondary structure, name all of the secondary structure elements and specify their positions in terms of the respective exterior and interior base-pairs. [10 marks]
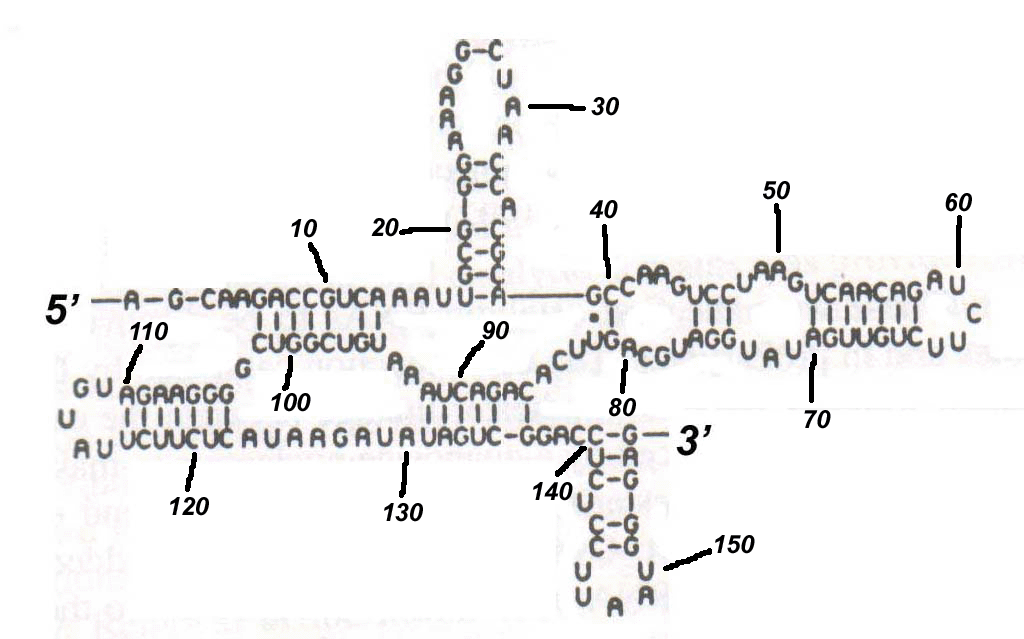
(b) Make structure from part (a) pseudoknot free by removing a minimal number of base pairings. (You don't have to redraw the structure, just list the positions of the base pairings that need to be removed.) [5 marks]
(c) Calculate free energy of the following RNA secondary structure based on the Turner Group energy parameters that can be found at https://www.bioinfo.rpi.edu/~zukerm/rna/energy/, use tables for 37 degree Celsius for RNA folding. [15 marks]
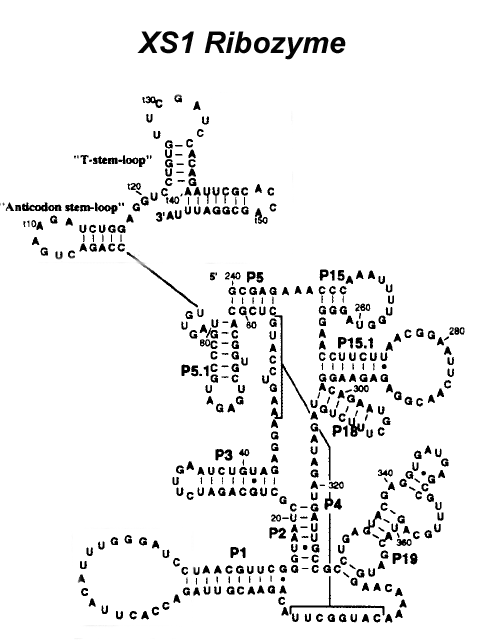
(a) Given the following sequence of the pseudoknotted XS1 ribozyme, use the MFOLD program of Zucker et al. to predict secondary structure (using default parameters), and compare the optimal structure to the original biological structure given in Figure 3. What do you notice? [5 marks]
XS1 ribozyme sequence and structure:
GCGAGAAACCCAAAUUUUGGUAGGGGAACCUUCUUAACGGAAUUC
AACGGAGAGAAGGACAGAAUGCUUUCUGUAGAUAGAUGAUUGCCG
CCUGAGUACGAGGUGAUGAGCCGUUUGCAGUACGAUGGAACAAAA
CAUGGCUUACAGAACGUUAGACCACUUACAUUUGGGAUCCUAACG
UUCGGGUAAUCGCUGCAGAUCUUGAAUCUGUAGAGGAAAGUCCAU
GCUCGCACGGUGCUGAGAUGCCCGUAGUGUCCAGACUGAAGAUCU
GGAGGUCCUGUGUUCGAUCCACAGAAUUCGCACCAGCGGAUUUA
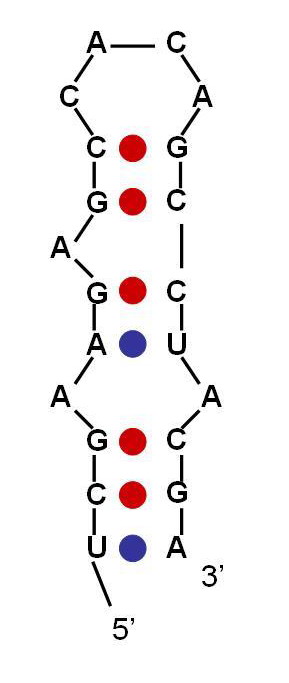
(b) Using the RNA Designer program of Andronescu et. al, design an RNA sequence that folds into the following structure given in dot-parenthesis notation (where dots represent unpaired bases and matching parenthesis indicate base pairs):
((((((.((.(((((((((........(((....))).......)))))..))))..)))))))).
Report the sequence and free energy obtained. [5 mark]
(c) Fold the following biological sequence using MFOLD (using default parameters):
AAACAGAGAAGUCAACCAGAGAAACAGACGUUGUCGUAUAAUACCUGGUACUGACAGUCCUGUUUU
What do you notice when you compare the free energy of the resulting structure and the minimal free energy from ten sequences for the same structure obtained from RNA Designer (with default parameters)? [10 mark]