(a) Show all steps of the UPGMA algorithm as applied to the following five sequences, where the distance between two sequences is defined as the number of base positions in which they differ. [10 marks]
(b) What is the Jukes-Cantor distance and why may it be more
appropriate than the simple distance measure used in part (a)?
(<= 50 words, in your own words).
[5 marks]
Note: We are not asking you to compute Jukes-Cantor distances
for the sequences from part (a).
(c) Briefly describe the role of "arithmetic averaging" in UPGMA. (<= 50 words, in your own words) [5 marks]
(d) Prove that Equation (7.2) from Durbin et al. gives the correct distances dkl between a merged cluster Ck = Ci + Cj (where '+' denotes set union) and every other cluster Cl according to the general definition of distance between clusters as given in Equation (7.1). [10 marks]
(e) Briefly explain how
one may determine a plausible root for an unrooted evolutionary tree obtained from
a phylogenetic tree inference algorithm, such as neighbour joining
(<= 50 words, in your own words).
[5 marks]
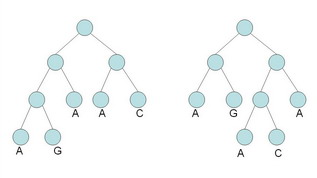
(a) Calculate the parsimony score of the two (rooted) trees shown below, using Fitch's algorithm. (Show all steps of your work.) What do you notice? [10 marks]
(b) Consider the left tree from part (a) with the sequences AC, GC, AG, AT, CG assigned to the leaves (from left to right). Solve the small parsimony problem for this tree. Show all steps of your calculation. You may reuse your results from part (a) as needed. [5 marks]
(c) Given n species and m characters
for each species, what is the time-complexity of solving the small
parsimony problem using Fitch's algorithm, assuming
that each set operation takes one time unit?
Explain your answer.
[5 marks]
Consider the following partial sequences from E.Coli clone vectors in FASTA format. (Source: https://www.cf.ac.uk/biosi/staff/ehrmann/tools/dnasequences.htm)
>pBR322
TTCTCATGTTTGACAGCTTATCATCGATAAGCTTTAATGCGGTAGTTTAT
CACAGTTAAATTGCTAACGCAGTCAGGCACCGTGTATGAAATCTAACAAT
GCGCTCATCGTCATCCTCGGCACCGTCACCCTGGATGCTGTAGGCATAGG
CTTGGTTATGCCGGTACTGCCGGGCCTCTTGCGGGATATCGTCCATTCCG
>pBR325
aggccatgtttgacagcttatcatcgataagctttaatgcggtagtttat
cacagttaaattgctaacgcagtcaggcaccgtgtatgaaatctaacaat
gcgctcatcgtcatcctcggcaccgtcaccctggatgctgtaggcatagg
cttggttatgccggtactgccgggcctcttgcgggatatcgtccattccg
>pBR327
TTCTCATGTTTGACAGCTTATCATCGATAAGCTTTAATGCGGTAGTTTAT
CACAGTTAAATTGCTAACGCAGTCAGGCACCGTGTATGAAATCTAACAAT
GCGCTCATCGTCATCCTCGGCACCGTCACCCTGGATGCTGTAGGCATAGG
CTTGGTTATGCCGGTACTGCCGGGCCTCTTGCGGGATATCGTCCATTCCG
>pACYC184
GAATTCCGGATGAGCATTCATCAGGCGGGCAAGAATGTGAATAAAGGCCG
GATAAAACTTGTGCTTATTTTTCTTTACGGTCTTTAAAAAGGCCGTAATA
TCCAGCTGAACGGTCTGGTTATAGGTACATTGAGCAACTGACTGAAATGC
CTCAAAATGTTCTTTACGATGCCATTGGGATATATCAACGGTGGTATATC
>pHSG575
TGATGTCCGGCGGTGCTTTTGCCGTTACGCACCACCCCGTCAGTAGCTGA
ACAGGAGGGACAGCTGATAGAAACAGAAGCCACTGGAGCACCTCAAAAAC
ACCATCATACACTAAATCACTAAGTTGGCAGCATCACCCGACGCACTTTG
CGCCGAATAAATACCTGTGACGGAAGATCACTTCGCAGAATAAATAAATC
>pGEX2T
acgttatcgactgcacggtgcaccaatgcttctggcgtcaggcagccatc
ggaagctgtggtatggctgtgcaggtcgtaaatcactgcataattcgtgt
cgctcaaggcgcactcccgttctggataatgttttttgcgccgacatcat
aacggttctggcaaatattctgaaatgagctgttgacaattaatcatcgg
(a) Use ClustalW (https://ibi.zju.edu.cn/clustalw/) to obtain a multiple sequence alignment of these sequences, using the ClustalW weight matrix. Show the result in Phylip format. [5 marks]
(b) Use the Phylip DNA parsimony program (https://bioportal.bic.nus.edu.sg/phylip/) to calculate evolutionary trees from the alignment from part (a) using parsimony. Report the resulting tree(s). [5 marks]
(c) Repeat the procedure from parts (a) and (b), but this time, use the IUB weight matrix for the multiple sequence alignment. Report the resulting multiple sequence alignment (in Phylip format) and the tree(s) obtained from parsimony. Briefly discuss the differences between the trees obtained here and in part (b). [10 marks]