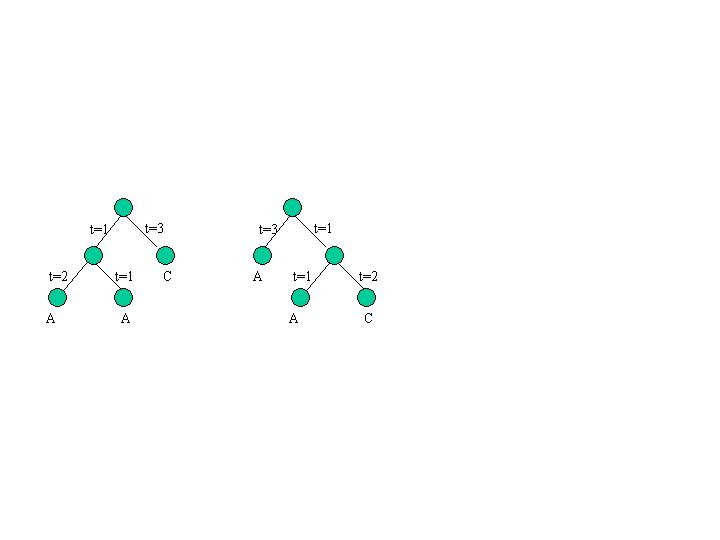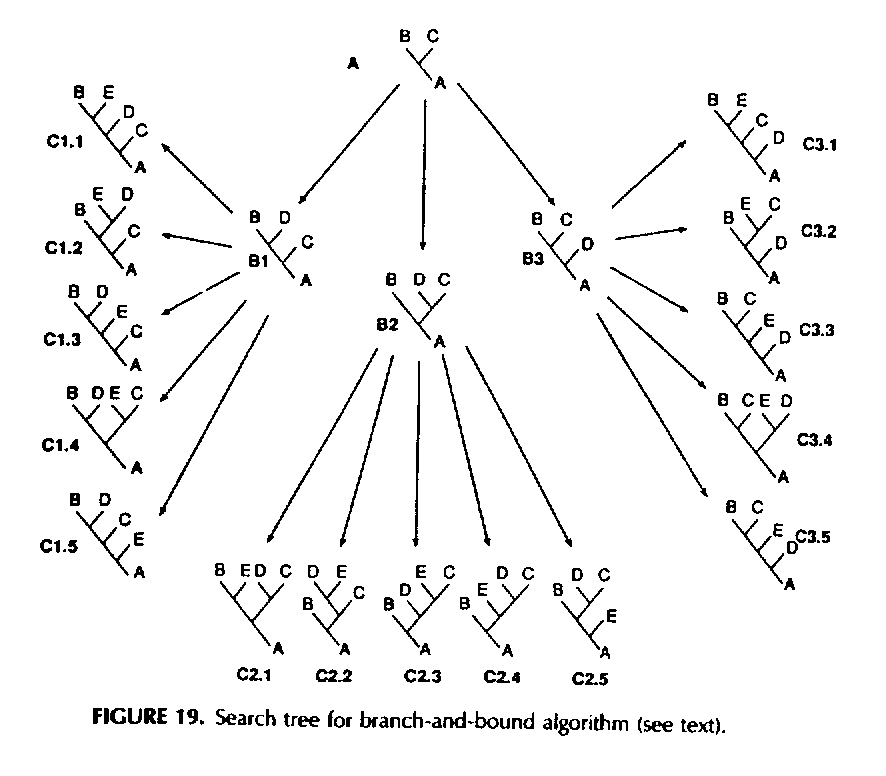
Following is an example of tree that can be explored by the branch and bound algorithm for finding an optimal parsimony tree. In this example, there are five input sequences, labeled A,B,C,D,E. Correspondingly, the leaves of the branch and bound tree (i.e. the nodes C1.1, C1.2, ..., C3.5) are labeled by possible phylogeny trees with five leaves. The root of the branch and bound tree (also, unfortunately, called A) is labeled by a phylogeny tree with three sequences. The internal nodes of the branch and bound tree, other than the root, (i.e. the nodes B1, B2, and B3) are labeled by phylogeny trees with four sequences.
Part (a): Suppose that the input sequences A,B,C,D, and E are each DNA sequences of length 1, with the following values: Sequence A and C correspond to the nucleotide "T" and sequences B,D, and E correspond to nucleotide "G". Score each of the trees A, B1, B2, B3, C1.1, ..., C1.5, C.2.2, and C.3.3 according to the (Fitch, unweighted) parsimony criterion.
Part (b): One "depth-first" ordering of the nodes of the
branch and bound tree is: A, B1, C1.1 C1.2, C1.3, C1.4, C1.5, B2,
C2.1, and so on, up to C3.5. Suppose that the branch and bound
algorithm explores the nodes of the branch and bound tree in this
order, keeping track of all of the best trees found at any point in
the exploration, and avoids exploration of nodes at level C of the
tree if their parent at level B of the tree already has a score that
is higher than the score of the best tree(s) already found. In this
case, list which nodes of the branch-and-bound tree are explored
by the branch and bound algorithm. Which trees are optimal?