- Phylogeny:
(evolutionary) relationships between any set of species
- Hypothesis: All organisms on Earth are evolutionarily related via a common ancestor
- Evidence:
similarity of many molecular mechanisms and genetic materials
- Phylogeny can be represented as a tree.
- 2 Types of Phylogenetics
- classic phylogenetics: based on morphological characters
- modern phylogenetics: based on information extracted from sequence data (DNA, RNA and proteins)
- based on characters = sites on sequences
- Tricky Issue: Gene Duplication
- common in nature
- leads to sequence divergence
- Paralogues vs. Orthologues
- Paralogues: genes diverged by gene duplication
- Orthologues: genes diverged by speciation
- Phylogenetic trees of species must be based on orthologues
- Assumption
- Sequences have descended from common ancestral genes/species,
but difficult to distinguish orthologues from paralogues - Phylogenetic tree of a group of sequences does not necessarily represent the true phylogenetic tree of host species
- Sequences have descended from common ancestral genes/species,
- leaves = species
- internal nodes = (hypothetical) ancestors
- nodes = species or character values (states)
- edges = evolutionary relationships between nodes
- edge lengths = evolutionary distance between nodes (evolutionary time)
- restrict ourselves to binary trees only
- ok, as we can use distances of 0
- rooted vs. unrooted trees
- root represents the ultimate ancestor of the group of sequences
(includes hierarchy)
- root represents the ultimate ancestor of the group of sequences
- n species
- m characters
- for each species, values for all characters
Want: fully labelled phylogenetic tree that 'best' explains the given data (i.e. maximize a target function (score) )
Assumptions:
- characters are mutually independent
- after two species diverged, their further evolution is independent of each other
Simple Solution: check them all out and pick the best one
- problem: too many possibilities to check
- n species -> (2n-3)!! different rooted trees
- n = 20 -> 1021 trees
- begin with a set of distances di between each pair i,j of seq.
- find the tree that predicts the observed sequence data as accurately as possible
Which distance metric?
- fraction f of sites u where xui /= xuj -> problem: doesn't 'correctly' model the distances for unrelated / highly diverged sequences don't want to distort evolutionary time
- better: probabilistic models of residue substitution e.g. Jukes-Cantor model for DNA dij = (-3/4)log(1 - 4f/3) ->dij -> infinity as f -> 0.75 (two totally random) for proteins, PAM matrices
How to find the tree
- general idea: given pairwise distance dij and tree T predicting
pairwise distance dij', look at:
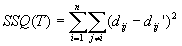
find the T that minimizes SSQ(T) => Least Squares Method
but NP-complete - Clustering: UPGMA (Unweighted Pair Group Method Using Arithmetic Averages)
- Idea: cluster sequences; at each stage, merge two groups and create a new node in the tree
- build the tree bottom up from the leaves
- distances dij of clusters Ci, Cj = average distance
between pairs of sequences from each cluster
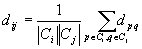
- complexity: polynomial
- result: rooted tree with molecular clock property (MCP)
- 1:1 correspondence between distance and evolutionary time
- not always true in reality; some sequences evolve faster
- if 'true' tree doesn't have MCP, UPGMA will give incorrect results
=> Yes, use neighbour joining - Neighbour Joining
- guarantees to generate correct tree in polynomial time if distance is additive
(weaker than MCP, so more reasonable; still, not always true) - Idea:
- find a pair of neighbouring leafs (in 'true' tree)
- remove then a set of leaves
- define the distance between the pair k and other other leaf m by
dkm = 1/2 (dim + djm - dij) - add k as a leaf
- iterate until done (i.e. only 2 leaves are left)
- can't just use the minimum distance between pair
- need to use correction factor
Dij = dij - (vi + ij)
(refer to Durbin et al., Chapter 7)
=>minimum Dij is guaranteed to represent neighbouring leaves in the true tree - Result: correct unrooted tree in polynomial time
- guarantees to generate correct tree in polynomial time if distance is additive