released: 2012/04/03, tue, 13:30; due: 2012/04/10, tue, 18:00 (via e-mail to Joel and Holger)
(a) Briefly outline the two different ways a motif can be defined formally that were covered in class (<= 50 words, in your own words). [4 marks]
(b) For each of the two definitions from part (a), briefly outline an algorithm for finding all instances of the motif in a given set of DNA sequences. [6 marks]
(c) Give the definition of relative entropy between two probability distributions. [1 mark]
(d) What is the smallest (absolute) value for the relative entropy of a distribution P w.r.t. another distribution Q? For which P and Q does this value occur? [2 marks]
(e) How is relative entropy used to evaluate candidate motifs? (<= 50 words, in your own words) [2 marks]
(f) Can the Gibbs Sampling Algorithm by Lawrence et al. find the maximum relative entropy motif for any given instance of the Relative Entropy Site Selection Problem with some probability > 0 in at most k iterations, where k is the number of sequences? Briefly justify your answer. [3 marks]
CATCATGACGTC
CTTCATGACGTG
GTGGATGACGTA
CACGAGGACGCC
CCGGATGACGCA
CCTAATGACGCA
GGGGATGACGTG
CACGATGACGTG
TTGGATGACGCT
GGTGATGACGTC
GCTCATGACGTA
TAGGATGACGCT
GGGGATGACGTC
CCTGATGACGAC
TCTAATGACGTA
TTCGATGACGTC
(a) Build a position weight matrix for the transcription factor CREB1.
[5marks]
(b) Write a program that computes the WMM scores for all windows (of the same length as the weight matrix). Run this program on the following sequence from the human genome chromosome 22:
AGCAGTATCAGGCACACTACCAGACCCAGGTAGAGACCGAGCACCTGCTG
GTGGGACATCAGCGACCATGCGACAGGGTCTGCCCAAGCGAGGTGTTGTC
AGATCCTCCAAGTCAGTAGGTCAGAGGCTCAGTAGCAATTGATGGTACAA
TGAAAAAGGAAGCCTTGGGCTTGGACCCAAGCGTGTCTAGTGGGTAACAG
TTATTTACAGGAAGAGGTGGCAGTGTGGCCCCACATCTGCTTTGCACTGA
TATTTCTCCCTTAGCAGATAAGCATTCTGGTCTGGTTCGTATATAGGTCA
GTTTGATGTGTTGATATGAACTGAAAATGAACCATAGGTACACTACAGTC
CCATTCAAGGTGACCCTAAAGGACGACAATAGGAAATCCTCCATGGGGCT
GAGCTTCCAGCAGTTCGTGTGGATATCCACTTTGTATGGAGGCAGTGGAC
AGAGTAGTGGCCTGAGGGAGGGACGCATATGGACTTCTGGGTTGTGACGT
CCTGCTGGCTGGTCAGGGACCTGAAAAGAGCAAGAGGGGAAGATGGACCT
ACAGGAGTGGCCACACAATATGTGCATCTCTCTGCCTTGTGTTAATACTG
CAGAGGAGTTGGTGAACAGCAGGATGGATGGGATGTCAGTCAGCTGTGCC
CCTGGCTACCCCTGTGCTTGAACAGTGGACTGTGAGTGGCGGCGTCATGC
AGAAGGAGCACAGGTTAGCGTCCACCAGCACAGGCCTTCTTTCTCAAGGC
TTGTCTCATGATTACTCCTGCTGAAAGCGATCAGTGCTGAGCCCCTGCTG
AGATACCATCCCTAGAGCACCCCAACTAGTTACTTAGTGGCAAGTTGGTG
ACAGCCCTTCATCCTTGCTGGAGTTGACACCTGCTCTGGATAAGGGTTTG
TCTTCTGTATCCACAGGGTCCCAGGCCAGCATCACTATCCAGGAGCTTCA
GGCATGCTCCGTGTACCAACATGGGATCACGAATCACATCGCCTCAGGCC
The length of this sequence is 1000 and, hence, there should be 989 windows for each the + and - strand directions. Write the window start positions (starting the position count at 0 for the first position in the sequence), the strand (+/-) and the scores to stdout (the console), such that each line contains a start position followed by a '+' or '-' followed by a score value, separated by a single space. Plot the scores (y axis) over window start positions (x axis) using any software that you like (GNUPlot, MS EXCEL, ..) and mark up (by hand or electronically) the regions that intuitively represent good hits. Hand in the graph with the rest of your written assignment. Note: The gene CHKB (chr 22) is known to be regulated by CREB1 transcription factor, but the CHKB gene is located on the reverse (-) strand. [35 marks]
Important notes:
|
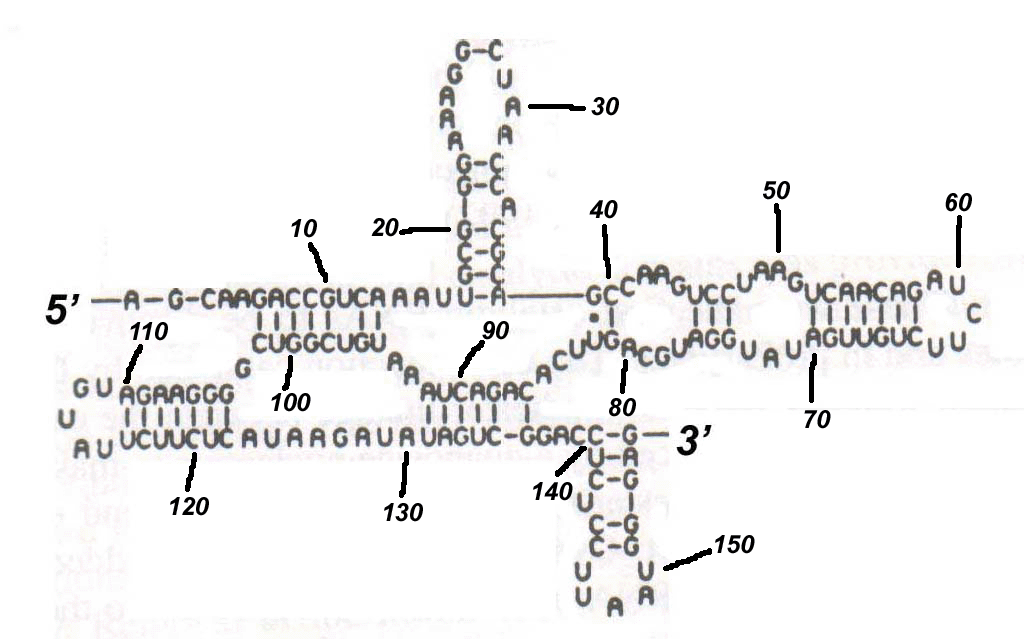
(a) Given the following RNA secondary structure, name all of the secondary structure elements and specify their positions in terms of the respective exterior and interior base-pairs. [10 marks]
Parts (b),(c) and (d) use the following sequence:
AACCCUUUCAAAAAGGGAGGUCACC
(b) Fill in the dynamic programming matrix produced from running the Nussinov algorithm on the following sequence (there are 6 blanks to fill in); [7 marks]
Nussinov score matrix:
| A | A | C | C | C | U | U | U | C | A | A | A | A | A | G | G | G | A | G | G | U | C | A | C | C | |
| A | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 2 | 3 | 3 | 3 | 3 | 4 | 5 | 6 | 6 | 6 | 6 | 7 | 8 | 8 | 8 | 9 | |
| A | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 2 | 3 | 3 | 3 | 3 | 4 | 5 | 6 | 6 | 6 | 6 | 7 | 8 | 8 | 8 | 9 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 3 | 4 | 5 | 6 | 6 | 6 | 6 | 7 | 8 | 8 | 8 | 9 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 3 | 4 | 5 | 5 | 5 | 5 | 6 | 6 | 7 | 7 | 8 | |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 3 | 4 | 4 | 4 | 4 | 5 | 5 | 6 | 6 | 7 | 8 | |
| U | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 5 | 6 | 6 | 6 | 7 |
| U | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 | 3 | 4 | 5 | 5 | 5 | 6 |
| U | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 | 5 | 5 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 2 | 3 | 3 | 4 | 5 | |||
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 4 |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 3 | 3 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 2 | 3 |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 2 | 3 |
| G | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 2 | 2 |
| U | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
(c) Given the following traceback report derived from the above matrix, fill in the empty labels as each step is popped from the stack as either paired, unpaired, bifurcation or NA. [5 marks]
| rows | columns | label |
| 1 | 25 | unpaired |
| 2 | 25 | unpaired |
| 3 | 25 | bifurcation |
| 3 | 17 | paired |
| 4 | 16 | |
| 5 | 15 | |
| 6 | 14 | |
| 6 | 13 | |
| 6 | 12 | |
| 7 | 11 | |
| 8 | 10 | |
| 9 | 9 | NA |
| 18 | 25 | |
| 19 | 25 | |
| 20 | 24 | |
| 21 | 23 | |
| 22 | 22 |
(d) Draw an optimal secondary structure for the sequence. You should use the work provided in (b) and (c). [3 marks]
Reading Questions: In graduate-style seminars, you will often be asked to read academic papers before class and have a set of questions prepared for discussion with your peers during the seminar period. These questions should not simply be limited to questions of the form, "I didn't understand X, how does it work?" but should demonstrate that you have made an effort to re-read and understand the paper to come up with more piercing, critical analysis, or suggestions for future improvements or directions to the work. Working out these questions will prepare you for such seminars and discussions in the future.
(a) Reading Question 1: Describe the method employed in "Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure" (http://bio.gsnu.ac.kr/research/miRNA/RNAss/Mfold/miRNAss_JMB_1999_Mathews.pdf) Why is the method an improvement at previous attempts at RNA Secondary Structure Predicition? When does the method have difficulty predicting structure accurately? What assumptions about the biological data does the method make?
(b) Reading Question 2: Read "Transat - A Method for Detecting the Conserved Helices of Functional RNA Structures, Including Transient, Pseudo-Knotted and Alternative Structures" (http://www.ploscompbiol.org/article/info%3Adoi%2F10.1371%2Fjournal.pcbi.1000823) and describe the method employed to predict functional RNA structures. Why is the method effective? When does the method have difficulty predicting structure accurately? What assumptions about the biological data does the method make?